Manipulation of Host Vesicular Trafficking and Membrane Fusion During Chlamydia Infection.
Manipulation of Host Vesicular Trafficking and Membrane Fusion During Chlamydia Infection
1. Introduction
Chlamydia infections are associated with a wide range of diseases. C. trachomatis (serovars A, B, Ba and C) causes trachoma, the world’s leading cause of infectious blindness. Serovars D through K are most commonly associated with sexually transmitted diseases and can cause infertility in women if left untreated [Paavonen and Eggert-Kruse, 1999]. Sexually transmitted diseases (STDs) are prevalent in every society in the world, including in developed countries (United States Centers for Disease Control [CDC]). Therefore, they represent a serious public health concern. Public programs aimed at increasing people’s awareness of the risks these pathogens pose have helped in controlling the spread of disease. Nevertheless, Chlamydia is still the most frequently reported STD in the United States. In 2009, 1.2 million new cases of Chlamydia infections were reported in the United States alone, but the actual number of infections is estimated to be higher due to a large number of unreported cases (CDC). Vaccination is the gold standard for disease prevention, but despite years of research, no vaccines exist for bacterial STDs. The typical course of treatment for Chlamydia infections involves the use of antibiotics, but there is emerging evidence that non-specific antibacterial agents can cause lasting damage to an individual by adversely affecting the homeostasis of the microbiota, which is the collection of bacteria that positively affect normal human functioning [Stewardson et al., 2011].
The Chlamydia life cycle consists of two distinct stages: the elementary body (EB) and the reticulate body (RB) stages. They are characterized by differences in morphology, metabolism, and infectivity [Tamura et al., 1971]. Elementary bodies are metabolically inactive but infectious (Figure 1-right). Extensive disulphide cross-linking of cysteine-rich outer membrane proteins allows the EBs to survive outside the host cell [Hackstadt et al., 1985]. Upon coming in contact with a host cell, the EB is endocytosed into a plasma membrane-derived phagosome in which it eventually differentiates into a larger, metabolically active (but non-infectious) organism called a reticulate body. The RB remodels the phagosome in a multi-step process that is still incompletely understood (see Figure 1- right). This remodeled phagosome is called an inclusion and is distinguished by the presence of bacterial virulence-associated proteins (also known as effectors) on its surface in addition to host proteins that are not normally found on typical phagosomes. The remodeling process involves the action of a Type 3 Secretion System (T3SS) [Stephens et al., 1998]. Pharmacological inhibition of T3SS proteins halts the translocation of certain bacterial proteins into the inclusion membrane [Muschiol et al., 2006].
Chlamydia express bacterial effectors on the inclusion, including proteins of largely uncharacterized function called inclusion proteins, or Incs [Li et al., 2008; Rockey et al., 1995; Scidmore and Hackstadt, 2001]. Inc family members are found across species of Chlamydiae but do not share a high level of sequence homology. They do, however, share several structural features including a characteristic bi-lobed N-terminal transmembrane domain [Bannantine et al., 2000] and a cytosolic C-terminal domain capable of interacting with host proteins [Rockey et al., 1997]. These interactions are discussed in further detail below. Inc proteins likely play a major role in bacterial pathogenesis. However, their exact roles in infection remain unclear, largely due to the lack of genetic techniques to easily and rapidly generate targeted knockouts of these genes.

Fig. 1. Comparison of Typical Phagosomal Maturation and Maturation of Chlamydia Phagosomes (Inclusions). (Left) A typical phagosome fuses with early and late endocytic compartments and gradually acidifies. Upon fusion with the lysosome, it becomes a phagolysosome and the bacteria inside are destroyed. (Right) Chlamydia elementary bodies are internalized into separate phagosomes. In the case of C. trachomatis, these phagosomes eventually fuse with each other into a large inclusion. Inside the cell, the Chlamydia inclusion interacts with early and late endocytic compartments, but avoids fusing with lysosomes. In addition, it promotes fusion with Golgi-derived vesicles, lipid droplets (LD), and multi- vesicular bodies (MVB) to acquire host nutrients. The Chlamydia inclusions do not greatly acidify as in typical phagosomes.
Chlamydiae possess a highly condensed genome consisting of just over 1 million bases. This genome lacks many genes and gene networks essential for metabolism [Stephens et al., 1998]. As a result, Chlamydiae are obligate intracellular bacteria and must adapt to thrive under extremely challenging conditions inside host cells. In particular, the pathogen has to co-opt host resources such as nutrients, proteins, or lipids for its own use without triggering immune defenses or killing the host. To this end, Chlamydiae have evolved sophisticated mechanisms to support their intracellular lifestyle. For example, Chlamydia inclusions appear to fuse with a select set of host vesicles while avoiding fusion with other organelles—namely lysosomes (Figure 1- Right) [Fields and Hackstadt, 2002]. Fusion with the host vesicles likely results in the delivery of vital bioactive compounds that the bacteria then use to support its growth and survival inside the cell. However, the metabolic pathways involved in the utilization of host compounds are incompletely understood. Inclusions do not maturate to phagolysosomes (Figure 1-Right), that is to say they do not display markers specific to lysosomes [reviewed in [Fields and Hackstadt, 2002]], in contrast to many non-pathogenic bacteria (Figure 1-Left). By inhibiting interactions with the endocytic pathway, Chlamydiae are able to evade destruction by innate immune defenses. How the bacteria accomplish this task is the subject of intense research.
Chlamydia infection is a complex process involving many factors. In this chapter, we will review data that suggest that Chlamydia inclusions selectively promote fusion with Golgi- derived vesicles, lipid droplets, and multivesicular bodies while avoiding fusion with the lysosomal compartments. We will look at how Chlamydia spp. remodel their inclusion using a Type 3 Secretion System and discuss new research that aims at understanding host vesicle re-trafficking at the molecular level.
2. The Chlamydia inclusion is a remodeled version of the phagosome
In healthy individuals, innate immune cells can recognize and destroy invading bacteria using phagocytosis [Metchnikoff, 1891]. Bacteria are rapidly internalized into a special organelle called a phagosome that successively fuses with early and late endosomes resulting in acidification of the phagosome. During the final step of maturation, the acidified phagosome fuses with lysosomes to become a phagolysosome (Figure 1-Left) [Botelho and Grinstein, 2011]. Inside the phagolysosome, bacteria are destroyed through the action of proteases, hydrolytic enzymes and toxic compounds such as reactive oxygen species [Botelho and Grinstein, 2011]. Chlamydiae however, avoid this degradative pathway altogether (Figure 1-Right). At no point during infection are lysosomal markers found on the inclusion membrane [Fields and Hackstadt, 2002]. This observation suggests that Chlamydia modify the inclusion membrane to render it undetectable to lysosomes thus allowing it to avoid fusion with those compartments and eventual acidification.
Chlamydia modify the phosphatidylserine content of the inclusion they inhabit One way to remodel the phagosomal membrane is by manipulating its lipid composition. A recent report looked at the localization of phosphatidylserine (PS) on phagosomes of inert and biologically active materials [Yeung et al., 2009]. Using a novel protein probe (referred to as Lact-C2) for membrane charge and lipid composition, the authors determined that PS contributes significantly to the lipid content of phagosomes. During Chlamydia infection however, Lact-C2 was found on the inclusion of C. trachomatis at only 6 hours post infection (hpi). At 18hpi, there was little or no Lact-C2 detected on the inclusion suggesting that C. trachomatis reorganized the lipid content of its inclusion. At this point, it is impossible to conclude with certainty what mechanisms are employed by the bacteria to alter the cellular distribution of PS.
In addition to remodeling the lipid composition of phagosomes, intracellular bacteria such as Chlamydia also modify the proteins on the membrane surface. It has been established that Chlamydia employs a Type 3 Secretion System to deliver bacterial proteins into the inclusion membrane and the host cytosol [Hueck, 1998; Subtil et al., 2001].
Secretion of Chlamydia proteins by a type 3 secretion system is necessary to protect the inclusion Type 3 Secretion Systems (T3SS) are multimeric bacterial needle-like superstructures found in many Gram-negative pathogenic bacteria. T3SS allow for the transport of effectors through the bacterial membranes and across a eukaryotic membrane [Worrall et al., 2011]. The genome sequence of C. trachomatis reveals three distinct gene clusters containing open reading frames (ORFs) encoding putative gene products that show significant homology to known T3SS proteins from Yersinia spp. These genes include the pore-forming complex YscC, the structural components of the inner membrane complex YscJ, YscR, YscS, YscT, and YscU, and the ATPase YscN and its binding partner YscL [Stephens et al., 1998] (Figure 2A). The Chlamydia protein CopB is found on the inclusion membrane and is believed to be a translocator operating as the gateway for effectors to enter the host [Peters et al., 2007].

Fig. 2. Chlamydia Type 3 Secretion System. (A) The Type 3 Secretion System (T3SS) shuttles proteins from the bacterial cytoplasm into the host cytosol or inclusion membrane. Shown here is a schematic depicting the transport of an inclusion protein from the bacterial cytoplasm where it is bound to a chaperone protein, to the inclusion membrane through the T3SS. OM = outer membrane (bacteria); IM = inner membrane (bacteria). (B) Schematic representation of a typical inclusion protein (Inc). The N-terminal peptide faces the cytosol. The bi-lobed transmembrane domain inserts twice into the bilayer, allowing the C-terminal peptide to face the cytosol as well.
Secretion systems are mostly known for their ability to inject soluble effectors into the host cytosol, however they are also capable of shuttling proteins containing hydrophobic transmembrane domains (Figure 2). The inability to genetically insert recombinant fusion proteins into Chlamydia has necessitated the use of heterologous systems to study proteins secreted by a Chlamydia T3SS. Dautry-Varsat and co-workers demonstrated that the inclusion membrane proteins IncA, IncB, IncC, along with Cpn0026, Cpn0146, Cpn0308, Cpn0367, and Cpn0585 from C. pneumoniae are transported through a T3SS in Shigella flexneri [Subtil et al., 2001]. The N-terminal domains of these proteins were fused to a cya (adenylate cyclase) reporter gene and secretion was measured by western blotting supernatant and pellet fractions. These experiments demonstrated that the secretion signal was encoded in the N-terminal region of the Chlamydia protein. Interestingly, all of the proteins mentioned above with the exception of IncB and IncC contain N-terminal bi-lobed transmembrane domains. This led to the speculation that the secretion signal may be located in that region (Figure 2B). Follow-up studies using the same heterologous system identified fifteen C. trachomatis proteins shown to localize to the inclusion membrane (as measured by immunofluoresence microscopy) and to be secreted by the S. flexneri T3SS [Dehoux et al., 2011].
The sequencing of the C. trachomatis genome [Stephens et al., 1998] allowed Chlamydia research to enter the bioinformatics age. One of the first efforts to predict inclusion membrane proteins (Inc) used the bi-lobed transmembrane domain architecture as a criterion for localization to the inclusion [Bannantine et al., 2000]. This was based on the observation that IncA, IncB, and IncC each contained this unusual motif and were identified as being on the inclusion membrane. The authors of this study found forty-six ORFs in the

This concept was extended when Zhong and colleagues investigated the intracellular location of fifty putative C. trachomatis inclusion membrane proteins [Li et al., 2008]. Antibodies were raised to putative Incs fused to either red fluorescent protein (RFP) or glutathione-S-transferase (GST). These antibodies were later used to confirm localization to the inclusion membrane. In total, twenty-two proteins were visible on the inclusion, seven were located within the inclusion, and twenty-one were undetectable by immunofluorescence.
Further evidence for a T3SS in Chlamydia has come in the form of pharmacologic inhibition of putative secretion-associated proteins. The compound INP0400, an inhibitor of the T3SS in Y. pseudotuberculosis, was shown to inhibit Chlamydia growth in cell lines when administered early during infection. For example, there was a decrease of the accumulation of 14-3-3b, a host phosphoserine-binding protein, around the inclusion, indicative of little or no IncG reaching the inclusion membrane [Muschiol et al., 2006]. The 14-3-3 family of proteins includes numerous members implicated in a myriad of pathways including apoptosis and tumor suppression [Morrison, 2009]. When INP0400 was added to cell culture media during the mid-cycle phase of infection, IncA function was apparently abrogated as multiple small inclusions were observed. The occurrence of multiple small inclusions (as opposed to one large inclusion) is indicative of a defective IncA protein [Fields et al., 2002; Hackstadt et al., 1999; Suchland et al., 2000]. The loss of functional IncA was attributed to a loss of translocation to the inclusion membrane as a result of the compound [Muschiol et al., 2006]. These findings are significant because they demonstrate the existence of an INP0400- sensitive protein or protein complex in Chlamydia likely to be a T3SS component.
Inclusion proteins can be post-translationally modified by the Host
The study of Chlamydia infection has largely focused on gross morphological changes during bacterial challenge. Re-organization of certain host markers and/or organelles implicates secreted bacterial effectors as being responsible for the observed phenotype. More recently, the role played by the inclusion in these phenomena has become more apparent. A complete identification of the inclusion proteome has still not been achieved. This could be due to the fact that (1) the proteome is likely to be dependent on the species of Chlamydia, the host cell or both; (2) it is dynamic and changes with length or phase of infection; (3) it is subject to the sensitivity and limitations of the techniques used to study it. Nevertheless, we can learn some basic facts about Chlamydia-host interactions through the study of known inclusion proteins. IncA and IncG for example, are two relatively well-studied proteins, each with identified binding partners. Importantly, discoveries regarding these two proteins have provided a starting point for the study of other inclusion proteins.
IncA was the first Chlamydia protein found on the inclusion membrane [Rockey et al., 1995]. Since then, IncA has been studied in relative depth. Microinjection experiments demonstrated that the soluble domain of IncA from C. psittaci is exposed to the host cytoplasm and that a host kinase is responsible for phosphorylating IncA [Rockey et al., 1997]. The function of IncA phosphorylation is unclear. C. trachomatis IncA is not known to be phosphorylated and no kinase for C. psittaci IncA has been identified [Fields and Hackstadt, 2002]. Furthermore, C. trachomatis IncA-mediated inhibition of endosomal fusion (discussed in detail below) was not reported to depend on phosphorylation [Paumet et al., 2009]. In contrast, C. caviae IncA was shown to be phosphorylated at serine 17, and this post
translational modification was required for the inhibition of C. caviae inclusion development in cell lines [Alzhanov et al., 2004]. These differences, though not mutually exclusive, highlight the biological diversity among Chlamydiae.
IncG is another Inc protein phosphorylated by the host [Scidmore and Hackstadt, 2001]. A yeast two-hybrid screen using C. trachomatis IncG as a bait revealed that the phosphoserine- binding protein 14-3-3b binds the C-terminal region of IncG. Green fluorescent protein (GFP)-tagged 14-3-3b localized to the inclusion membrane in infected HeLa cells thus confirming the physiological relevance of this interaction. Serine 166 was determined to be a phosphorylation site as a mutation of this residue to alanine changed the electrophoretic mobility of IncG, consistent with a change in phosphorylation state. The S166A mutation abolishes interaction between IncG and 14-3-3b in a yeast two-hybrid assay. As for IncA, the kinase responsible for phosphorylating IncG is unknown. That both yeast and human cells apparently phosphorylate the same residue on IncG strongly suggests that the kinase is conserved from yeast to humans. Of course, the data do not rule out the possibility of multiple kinases being able to modify IncG.
Why Chlamydia would need to traffic 14-3-3b to the inclusion membrane is unknown. One possibility is that it helps form a multi-protein complex at the inclusion-cytoplasm interface. Another possibility is that IncG sequesters 14-3-3b to the inclusion and prevents it from functioning in its normal location in the cell. In any case, the finding that a Chlamydia protein is able to interact with 14-3-3b is significant because it implicates a connection between the inclusion and host cell cycle regulation. It should be pointed out that the interaction between 14-3-3b and IncG appears specific to C. trachomatis as the antibodies used to probe IncG on the inclusion of C. trachomatis strains A, B, Ba, C, D, E, F, G, H, I, J, K, L1, L2, and L3 did not cross-react with that of other species (namely C. psittaci and C. pneumoniae) [Scidmore and Hackstadt, 2001].
To date, phosphorylation is the only post-translational modification detected on Inc proteins. Additional modifications, such as glycosylation, would certainly be interesting, as it would extend the variability and functions of the bacterial effectors and possibly highlight new pathways manipulated by Chlamydiae. As molecular biology techniques become more sophisticated and comprehensive, we will be able to better probe these types of questions in the future.
In addition to modifying the phagosome they inhabit with the insertion of bacterial proteins through the T3SS, Chlamydia also manipulate the host proteins and lipids in order to change the nature of this compartment. They accomplish these modifications by (1) hijacking vesicles from the Golgi apparatus, (2) fusing with the multivesicular bodies and (3) corrupting host lipid droplets (see Figure 1-Right, and the following sections for more details).
3. Evidence for the fusion of Golgi vesicles with the inclusion membrane The Golgi apparatus is the site of intracellular sorting of cargo. A variety of proteins— including members of the Rab and SNARE families—regulate trafficking between the Golgi and the different intracellular compartments or the plasma membrane [Conibear and Stevens, 1998]. The specific distribution of these proteins ensures a high degree of processivity and organization in cargo sorting [McNew et al., 2000; Paumet et al., 2005;
Paumet et al., 2004]. Perturbations of this network result in aberrant localization of cargo to incorrect compartments. One hallmark of Chlamydia infection is the disruption of vesicular trafficking and the redirection of exocytic vesicles to the inclusion [Hackstadt et al., 1995; Heinzen et al., 1996]. The delivery of lipidic cargo to the inclusion likely gives Chlamydia access to nutrients required for its intra-inclusion survival. In addition, this may give the inclusion a Golgi-like identity, helping it to escape the degradative pathway. However, there is experimental evidence showing that withholding lipids does not starve and/or kill the pathogen, but rather destabilizes the inclusion thus weakening the infection [Robertson et al., 2009].
Host-derived sphingomyelin is a key component of Chlamydia development Direct evidence for an interaction between the inclusion and Golgi vesicles first came in 1995 when a derivative of the fluorescent Golgi marker {N-[7-(4-nitrobenzo-2-oxa-1,3- diazole)]}aminocaproylsphingosine (C6-NBD-ceramide) was shown to localize to the inclusion in a brefeldin A-sensitive manner [Hackstadt et al., 1995]. Brefeldin A inhibits cis-to-trans Golgi trafficking. Interestingly, Brefeldin A abrogated the association of the C6-NBD-ceramide with the inclusion leading to the conclusion that Chlamydiae intercept exocytic vesicles coming from the trans-Golgi network as opposed to the cis-Golgi face. The lipid species in question was determined to be sphingomyelin based on thin layer chromatographic analysis of purified inclusions. Later, Beatty and colleagues discovered that chemical inhibition of sphingomyelin acquisition in infected cells led to a corresponding loss of inclusion membrane integrity characterized by the absence of homotypic (inclusion-inclusion) fusion. In addition, they observed the premature differentiation from RBs to EBs, their early release, and a decrease in the ability of the bacteria to reactivate after reaching a persistent infection [Robertson et al., 2009]. Next, the authors inhibited host sphingomyelin synthesis at different steps during the metabolic pathway and analyzed the effects these compounds had on Chlamydia development. They found that pharmacologic inhibition by myriocin, which inhibits serine palmitoyltransferase (SPT) [Johnson et al., 2004], or fumonisin B1, which inhibits sphingosine N-acyltransferase downstreatm of SPT [Schroeder et al., 1994], correlated with the presence of multiple small inclusions in the cells tested [Robertson et al., 2009]. Addition of either dihydroceramide or sphingosine (two metabolites whose levels are negatively affected by myriocin) to the culture media restored normal inclusion morphology, validating that host sphingolipid synthesis is required for normal inclusion development. Chlamydia grown in cell lines with a nonfunctional SPT display defects similar to myriocin-treated cells. Complementation of the SPT proteins restores normal bacterial growth and development. Interestingly, subversion of Golgi-derived vesicles may actually occur through the hijacking of multivesicular bodies (MVBs) as treatment of infected cells with the MVB inhibitor U18666A disrupted the progression of the inclusion and produced phenotypes similar to that seen with myriocin treatment and SPT depletion [Robertson et al., 2009]. We discuss the manipulation of the MVBs by Chlamydia in section 4. In summary, these data strongly support that host- derived lipids, particularly sphingomyelin, are essential for Chlamydia. Interferring with this process results in apparently dysfunctional inclusion membranes.
Acquisition of Golgi-associated Rab proteins by the inclusion
Rab proteins are members of a large family of proteins (Ras-like proteins) conserved across eukaryotes. They regulate many aspects of membrane trafficking from specificity and tethering to fusion [Hutagalung and Novick, 2011]. Rabs are found in either a soluble or
membrane bound form (Figure 3). They are attached to membranes via lipid anchors. In addition, they can be found in an “inactive” (GDP1-bound) or “active” (GTP2-bound) state. The activation state of Rabs is controlled by GEPs (guanine nucleotide exchange proteins) and GAPs (GTPase activating proteins). GEPs exchange GDP for GTP thus setting Rabs to an “active” state, whereas GAPs cause Rabs to hydrolyze the bound GTP to GDP causing them to enter an “inactive” state [Pfeffer, 2001; Segev, 2001].
During Chlamydia infection, Rabs involved in Golgi membrane fusion are found on the inclusion. Scidmore and colleagues analyzed the subcellular localization of eight Rabs (Rab1, 4, 5, 6, 7, 9, 10, 11) in cells infected with Chlamydia and found that the recruitment of specific Rabs to the inclusion was dependent on the species of Chlamydia [Rzomp et al., 2003]. During C. trachomatis infection, overexpressed Rabs1, 4, 6 and 11 relocate to the inclusion membrane as visualized by fluorescent microscopy [Rzomp et al. 2003]. The association between Rab6 and the inclusion appears specific to C. trachomatis since Rab6 was not found on the inclusion when cells were infected with either C. muridarum or C. pneumoniae. Rab10 however, seemed to localize only to the inclusions of C. muridarum and C. pneumoniae but not to that of C. trachomatis. Interestingly, the presence of Rabs on the inclusion was not dependent on intact microtubules, as inhibition of microtubules by nocodazole did not affect Rab acquisition.

Fig. 3. Rab Proteins Regulate Membrane Fusion Through GTP Hydrolysis. (Left) Rabs are inactive when bound to GDP. A network of regulatory proteins coordinates activation or inhibition of Rab activity. GTP Exchange Proteins (GEPs) replace bound GDP with GTP and activate the Rab protein (Right). GDP Dissociation Inhibitors (GDIs) keep the Rab bound to GDP and locked in the inactive state. Once activated, Rab GTPases recruit effector proteins that act on downstream targets to promote membrane tethering and fusion. Afterwards, a GTPase Activating Protein (GAP) stimulates the GTPase activity of the Rab GTPase, which in turn hydrolyzes bound GTP to GDP and enters an inactive state.
Of the proteins found to associate with the inclusion, Rabs1, 6, and 10 are intimately involved in Golgi trafficking. Another Golgi Rab protein, Rab14, was later shown to localize to the C. trachomatis inclusion [Capmany and Damiani, 2010]. This localization was dependent on prenylation of Rab14 and occurred approximately 10-18 hours post-infection. Blocking bacterial protein synthesis with chloramphenicol treatment ablated the association between the inclusion and Rab14 suggesting that inclusion proteins are involved in Rab14 acquisition. IncA is detectable approximately 10-12 hours post infection and does not seem to be the main effector for Rab14 acquisition because incubation of infected cells at 32°C, which is known to block IncA expression on the inclusion surface [Fields et al., 2002], did not adversely affect the detection of Rab14 on the inclusion [Capmany and Damiani, 2010]. Notably, expression of the dominant negative (soluble) form of Rab14 inhibited Chlamydia growth most likely by blocking transport of sphingolipids to the inclusion.
The recruitment of Rab proteins likely acts to facilitate selective fusion between the inclusion and other intracellular compartments. One possible mechanism is the formation of tethering complexes that tie the inclusion to another vesicle or organelle. Rabs have been shown to be present in tethering complexes mediated by the formation of scaffolding complexes or by the action of coiled-coils [Hutagalung & Novick 2011]. It is interesting to note that several inclusion proteins are predicted to form coiled-coils. However, their structures are yet to be defined.
SNARE proteins are recruited to the inclusion membrane
The proteins located on the surface of the inclusion membrane, including both bacterial effectors and host proteins, are unique because of their position at the interface between the host and the pathogen. The abnormal composition of the Chlamydia inclusion membrane is key to survival and a necessity for pathogenesis. In this section, we will look at the recruitment of specific host SNARE proteins to the inclusion.
SNAREs are ubiquitously expressed proteins that mediate intracellular membrane fusion and are conserved from yeast to humans [Bock et al., 2001; Pelham, 1999]. They constitute the core machinery necessary to mediate specific membrane fusion [McNew et al., 2000; Parlati et al., 2002; Paumet et al., 2001]. As such, they are key regulators of all intracellular vesicle trafficking events and cargo transport steps. These proteins, present on the surface of almost all intracellular compartments [Chen and Scheller, 2001], assemble into a stable complex. As a result, both membranes are brought in a close apposition until fusion occurs (Figure 4). SNAREs are divided into two broad categories depending on their relative locations: target- (t-SNAREs) and vesicle- (v-SNAREs) SNAREs. Whereas the v-SNARE is always a single protein, the t-SNARE is composed of three subunits, one heavy chain and two light chains [Fukuda et al., 2000]. Notice that both light chains can be encoded in one or two separate proteins. A typical SNARE protein encodes a coiled-coil SNARE domain containing a heptad repeat motif spanning approximately 70 amino acids, and a C-terminal site that anchors it to a membrane. The SNARE domain contains a polar or charged residue located in the middle of the domain (also known as “zero-layer” [Fasshauer et al., 1998]) (see Figure 5A). The C-terminal anchoring site can be a simple hydrophobic transmembrane domain or a palmitoylation site.
Fusion specificity is largely encoded in the partnering of cognate SNARE proteins [McNew et al., 2000; Parlati et al., 2002; Paumet et al., 2001; Paumet et al., 2004]. Altogether, their characteristics designate SNAREs as excellent markers to determine the identity and activity of individual compartments in intracellular transport. Therefore, manipulation of individual SNARE proteins or of the complex itself may help intracellular pathogens to subvert normal trafficking patterns. As such, the recruitment of host SNAREs would afford the pathogen the ability to manipulate vesicular trafficking during infection.

Fig. 4. Diagram of SNARE-Mediated Fusion. (A) In resting cells, SNARE proteins interact with effector proteins that control their availability. (B) Following a stimulatory signal, the t- SNARE proteins interact to form a complex that in turn binds the v-SNARE. As a result, a trans-SNARE complex forms (four-helix bundle) between t-SNAREs on the target membrane and a v-SNARE on the vesicle. (C) As the SNARE complex zippers from N- to C-terminal, the space between the opposing membranes decreases. (D) Fusion occurs when both bilayers merge and form a fusion pore. Following fusion, the trans-SNARE complex becomes a cis-SNARE complex (so called because all the SNAREs are now located on the same membrane). (E) The cis-SNARE complex is disassembled by NSF, an ATPase bound to its co-factor a-SNAP at the level of the SNARE motifs. (F) ATP hydrolysis dissociates the four-helix bundle, and the SNARE proteins are trafficked back to their original cellular compartments.
Syntaxin 6 is a SNARE protein that localizes to the trans-Golgi network (TGN) and likely plays a role in mediating the fusion of Golgi-derived vesicles with endosomes and/or lysosomes [Bock et al., 1997]. Recently, Hackstadt and co-workers showed that Syntaxin 6 relocates to the inclusion in a 18 hours post-infection [Moore et al., 2011]. Neither Syntaxin 4 nor Syntaxin 16 were found to localize to the inclusion suggesting that the recruitment of Syntaxin 6 was specific. Furthermore, the re-location of Syntaxin 6 to the inclusion is conserved among several species including C. trachomatis, C. muridarum, C. caviae, and C. pneumoniae. Treatment of the infected host cells with chloramphenicol to inhibit bacterial
protein synthesis abrogated trafficking of Syntaxin 6 to the inclusion suggesting that bacterial effectors are necessary to induce this recruitment. The depletion of Syntaxin 6 by siRNA did not significantly affect the development of the inclusion nor change the recruitment of sphingomyelin to the inclusion [Moore et al., 2011]. Therefore, the role f Syntaxin6 during Chlamydia infection remains unclear.
SNARE domains mediate protein-protein interactions between SNAREs and are required for fusion. Interestingly, the SNARE domain of Syntaxin 6 was not required for the recruitment to the inclusion. Instead, a small 10-amino acid “plasma membrane retrieval sequence” (TDRYGRLDRE) located N-terminal to the SNARE domain was found to be necessary for re-localization [Moore et al., 2011]. Deletion of this domain results in Syntaxin 6 being present in the cytosol in non-infected cells [Watson and Pessin, 2000].
Perspectives on Golgi fusion
These observations bring up an important, and as yet unanswered, question: how do Chlamydia inclusions interact with exocytic vesicles to acquire sphingomyelin and presumably other lipids? The process apparently involves an energy-dependent step. Depletion of adenosine triphosphate (ATP) using 2-deoxyglucose and sodium azide significantly lessened the amount of fluorescent C6-NBD-ceramide found on the inclusion [Hackstadt et al., 1996]. Incubation of cells at 20°C, which would presumably stop active vesicle fusion, also resulted in fewer C6-NBD-ceramide molecules observed on the inclusion membrane. These data lead to the attractive hypothesis that members of the eukaryotic membrane fusion machinery (i.e. SNAREs, Rabs) are involved in lipid acquisition. This possibility is supported by the fact that Syntaxin 6, a TGN SNARE is specifically recruited on the inclusion and could potentially be involved in the hijacking of the Golgi vesicles. The extent to which SNAREs play a role in Chlamydia pathogenesis is unclear, but evidence is mounting that the interaction between effectors and SNAREs (or the inhibition of certain SNAREs by the bacteria) is an important hallmark of infection.
4. Multivesicular body markers are recruited to the Chlamydia inclusion Multivesicular bodies (MVBs) are endosomal compartments that are involved in the sorting of surface proteins to lysosomes. They get their name from the multiple intralumenal vesicles that form as the result of invagination events at the MVB membrane. Proteins destined for destruction by lysosomal degradation are mono-ubiquitinated within the MVB and cluster at the invagination sites so that they can be exposed to degradative proteins of the lysosome for destruction.
It was traditionally thought that the Chlamydia inclusion did not intersect with the endosomal pathway. However, it appears that inclusions interact with MVBs and recruit MVB-associated proteins to their surface. In a series of microscopy studies, Beatty and colleagues identified three MVB markers—CD63, lysobisphosphatidic acid, and metastatic lymph node 64 (MLN64)—that trafficked to the inclusion after initial infection [Beatty, 2006]. Also observed was the absence of LAMP-1 (lysosome-associated membrane protein 1). This is in agreement with previous observations concerning the selective recruitment of vesicle- associated membrane proteins (see Introduction). CD63 actually appeared to be both on the inclusion membrane and inside the inclusion. Cryo-electron microscopy confirmed this finding [Beatty, 2006]. Lumenal CD63 seemed closely associated with the bacterial cell wall. Despite its close proximity with the bacteria, CD63 does not appear to play a significant role in the life cycle of C. trachomatis since the ablation of CD63 by small interfering RNA (siRNA) does not affect normal Chalmydia growth [Beatty, 2008].
The role of MVBs in Chlamydia infection is incompletely understood. One possibility is the acquisition of host lipids such as sphingomyelin [Beatty, 2008; Robertson et al., 2009]. The discovery that CD63 is transported in the inclusion lumen is important because it hints at a mechanism for acquisition of MVB components. At this point it is pure speculation as to what is being imported or how it is getting there. The most basic explanation for this phenomenon is that MVBs are fusing with the inclusion membrane and the internal vesicles are released into the lumen. Indeed, CD63 has been found on the internal vesicles of MVBs (microvesicles) [Heijnen et al., 1999]. This raises the interesting possibility that the internal vesicles of MVBs may actually be the main targets of Chlamydia as a source of nutrients.
MVBs are not the only way that Chlamydia acquire new lipids. Recently, it has been demonstrated that these bacteria were also able to hijack and fuse lipid droplets with their inclusions.
5. Corruption of host lipid droplets by Chlamydia
Lipid droplets play a role in intracellular homeostasis Lipid droplets (LD) are intracellular stores of neutral lipids. LDs are found in a wide variety of eukaryotic cell types and are composed of both lipids and proteins. Once thought of as passive artifacts, LDs are now recognized as legitimate bioactive organelles. Disruption of lipid droplets (LDs) is associated with diseases such as type 2 diabetes and obesity [Farese and Walther, 2009]. It is believed that the initiation of LD formation begins with a swelling of the endoplasmic reticulum phospholipid bilayer caused by a gathering of neutral lipids. This is followed by a pinching off of the cytosol-facing leaflet to generate a cluster of neutral lipids, sterols, and sterol esters encased in a phospholipid monolayer. LDs are unique in the sense that they are the only recognized organelles to utilize a phospholipid monolayer. The exact function of LDs is unclear. Nevertheless, multiple interactions between LDs and other organelles suggest a physiologically significant role in cellular processes like metabolism.
Chlamydia proteins interact with host lipid droplets A yeast genetic screen of the Chlamydia ORFeome uncovered three bacterial proteins (called Lipid Droplet Associated proteins, or Lda) that co-localized with yeast lipid droplets as measured by immunofluorescence: CT156 (Lda1), CT163 (Lda2), and CT473 (Lda3) [Kumar et al., 2006]. These proteins were also shown to bind to LDs in mammalian cells. Interestingly, Lda1 and Lda3 can be stably expressed in HeLa cells, but Lda2 seems to require LD biogenesis to be stable and avoid degradation in vivo. EGFP:Lda (enhanced green fluorescent protein) fusion proteins localized to both LDs and the inclusions themselves [Kumar et al., 2006].
Lda2 is unstable in the absence of lipid droplets. Incubation of C. trachomatis-infected cells with the compound triacsin C, which inhibits synthesis of cholesterol esters and triacylglycerides, resulted in the formation of smaller inclusions than those found in non treated cells [Kumar et al., 2006]. It is not yet clear what role LD biogenesis and corruption plays in Chlamydia infection, but it appears to be a process critical for bacterial survival. Likewise, the exact association of Lda proteins with the membranes is unknown. Although none of the three proteins appear to contain the bi-lobed TMD structure commonly associated with inclusion proteins, it is possible that Ldas contain other as-yet- uncharacterized TMDs. It is also possible that Ldas are soluble and associate with host or Chlamydia proteins already present on the respective surfaces. Another compelling possibility is that post-translational modifications could lead to the addition of a lipid moiety that anchors the protein to the membrane surface. This matter is further complicated by the fact that no signal sequence specific for LDs has been identified.
Another interesting question is whether these Lda proteins are translocated through a Type 3 Secretion System. Secretion systems are a very efficient means of transporting proteins across biological membranes and are found in most, if not all, pathogenic bacteria studied to date (see section 2.1 of this chapter for more information on the Chlamydia T3SS). An active secretion apparatus would seem the most logical method of translocation because two lipid bi-layers separate the bacteria from the host cytosol (Figure 2A). The Ldas would have to traffic through these bilayers in order to gain access to lipid droplets. However, no T3SS screen has identified Lda proteins as being substrates for secretion so it is possible that Ldas are shuttled through another secretion system or by a novel method of translocation.
The lipid droplets themselves are translocated across the inclusion membrane into the lumen where they appear to physically associate with the reticulate bodies [Cocchiaro et al., 2008]. The mechanism by which LDs fuse with the inclusion membrane is unclear. LDs appear as whole, intact organelles throughout the process of translocation, providing evidence that the neutral lipids observed to be inside the inclusion are not the results of individual lipid molecules being trafficked across the membrane but rather bulk import of the entire droplet [Cocchiaro et al., 2008; Kumar et al., 2006]. A major unresolved issue is how the reticulate bodies interact with the lipid droplet once inside the inclusion lumen.
Lipid droplets are also capable of fusing with one another in a process that may resemble SNARE-mediated membrane fusion [Bostrom et al., 2007]. In this study, the authors found that SNAP23, Syntaxin 5, and VAMP4 associated with LD homogenate. In addition, NSF and a-SNAP, two proteins regulating SNARE complex dissociation, were also found to be associated with LDs via Western blot. This suggests that LD homotypic fusion might be SNARE-mediated. This conclusion is strengthened by the observation that inhibition of expression of any one SNARE (SNAP23, Syntaxin 5, or VAMP4) using siRNA constructs, significantly inhibited the rate of fusion of LDs.
6. The Chlamydia inclusion intersects with early and late endosomes but avoids fusion with the lysosome and acidification Chlamydia does not fuse with the lysosomal compartment Bacteria that are detected by host innate immune cells are endocytosed into phagocytic compartments that successively fuse with early and late endosomes resulting in the acidification of the phagosome and, ultimately, fusion with the lysosome where they are destroyed (Figure 1-Left). It has been observed that enzymes contained in the lysosome including acid phosphatase, arylsulfase, and b-acetylglucosaminidase do not enter the Chlamydia inclusion demonstrating a lack of fusion with this compartment [Todd and Storz, 1975]. How Chlamydiae block this process is incompletely understood, although new data have been gathered showing that Chlamydia is able to use SNARE-like proteins to block lysosomal fusion [Paumet et al., 2009; Wesolowski and Paumet, 2010] (see section 8). It remains unclear at which step elementary (or reticulate) bodies deviate from the lysosomal pathway. The situation is further complicated by the fact that certain markers for early and late endosomes are found on inclusions [van Ooij et al., 1997]. Yet, the inclusion itself does not acidify [Schramm et al., 1996].
The transferrin receptor and CI-M6PR associate with the inclusion at distinct stages of infection Chlamydiae have long been known to avoid lysosomal degradation (see Introduction). However, its association with other endocytic compartments has been observed. Engel and co-workers have examined the localization of early and late endocytic markers (transferrin receptor (TfR) and cation-independent mannose-6-phosphate receptor (CI-M6PR), respectively) with the inclusion as a function of time in HeLa cells infected with C. trachomatis [van Ooij et al., 1997]. What they observed was that, at approximately 4 hours after infection, antibodies against the TfR and CI-M6PR3 stained brightly around the inclusion indicating possible fusion between the inclusion and early and late endosomes. These associations remained for up to 20 hours post-infection. Interestingly, the staining patterns were different for both proteins at 4 and 20 hours post-infection. At the later time point, a more defined membrane staining of the inclusion was observed as opposed to an aggregation-like pattern seen at 4 hours. Further interaction between TfR-containing endosomes and inclusions was observed when looking at the rate of transferrin recycling in C. trachomatis-infected cells. Recently, the association of endocytic SNAREs including VAMP3, VAMP7 and VAMP8 has been identified [Delevoye et al., 2008], supporting the interaction between the Chlamydia inclusion and the endocytic pathway.
Wyrick and co-workers used a pH-sensitive fluorophore attached to elementary bodies of C. trachomatis to measure the relative pH of compartments in which the bacteria reside. They discovered that despite intersecting with late endosomes, the inclusion does not reach a pH lower than 6 [Schramm et al., 1996]. Also, the pharmacologic inhibition of Na+/K+ ATPases, which oppose acidification of endosomes due to vacuolar ATPases, inhibits Chlamydia growth presumably due to acidification of the phagosome [Schramm et al., 1996]. Inhibition of v-ATPases had no effect on bacterial growth. This is in agreement with microscopy data showing that v-ATPases are not found on inclusion membranes and that the compounds chloroquine and ammonium chloride (which raise the endosomal pH) had no effect on Chlamydia growth [Heinzen et al., 1996]. In addition to v-ATPases, neither LAMP1 nor LAMP2 were found on the inclusion membrane in the study.
In summary, although Chlamydia inclusions do interact to some extent with the endocytic pathway, they actively exclude certain endosomal markers—particularly those corresponding to lysosomes. Enzymes associated with lysosomal compartments were also apparently excluded from the inclusion lumen suggesting selective inhibition of fusion.
7. Chlamydia use SNARE-like proteins to block SNARE-mediated lysosomal fusion
The lack of a genetically pliable system for Chlamydia is a major obstacle in the study of the pathogen. However, scientists are finding new and innovative ways to study bacterial proteins in vitro. Inclusion-bound bacterial proteins are poorly characterized in terms of structure and function. Much research has focused on determining which proteins traffic to the inclusion, but their respective roles in infection are unknown. Inc proteins share very little sequence homology to any known eukaryotic proteins. Even within the same species of Chlamydia, the sequence homology of these proteins is very low. Importantly, all known members of Chlamydiae possess proteins localizing to the inclusion suggesting a critical role in survival. Recently, we have made progress in understanding how these proteins interact with host proteins. In particular, the interaction between IncA and members of the host membrane fusion machinery—namely SNARE proteins—have been indentified. The mechanism(s) uncovered in these types of studies will shed new light on the process by which Inc proteins help to subvert vesicle trafficking and establish a replicative niche inside the host.
The SNARE domain is a specialized coiled-coil domain
The defining feature of SNARE proteins is the presence of one or more SNARE domains. SNARE domains are around 70 amino acids in length and consist of tandem heptad repeats of the form A-B-C-D-E-F-G where A and D are hydrophobic residues and B, C, E, F, and G are polar or charged [Antonin et al., 2002; Sutton et al., 1998] (Figure 5A). In the SNARE domain, the A and D residues are known as layers and form tight hydrophobic interactions. Within the SNARE domain, there is a single polar or charged residue that resides in the D position towards the middle of the domain. This position is known as the 0-layer (zero-layer) and is discussed in detail below. In the vast majority of SNARE proteins, this residue is either a glutamine or an arginine4 [Fasshauer et al., 1998]. It has been proposed that the formation of the four-helix bundle begins at the N-terminus of the SNARE domain and proceeds straight through the C-terminal transmembrane domains [Melia et al., 2002; Pobbati et al., 2006]. This zipper-like motion pushes water molecules away from the hydrophobic core of the complex and brings the two membranes in close enough proximity to bring about fusion [Fasshauer, 2003; Melia et al., 2002; Stein et al., 2009]. This four-helix bundle assembles in a very organized and predictable way. The 0-layers of each SNARE domain align and interact in the final four-helix bundle as mentioned above. Likewise, each successive hydrophobic residue in the A and D positions will align with other hydrophobic residues on cognate SNAREs the same distance away from the 0-layer creating additional layers of hydrophobic interactions. Layers are numbered according to their distance from the 0-layer. In order to understand whether Chlamydia is using SNARE-like proteins to manipulate fusion, this characteristic SNARE domain was used as a matrix to identify similar domains in the Inc proteins [Delevoye et al., 2008].

Fig. 5. The SNARE Domain is Organized into Discrete Hydrophobic Layers. (A) The SNARE domain is a coiled-coil domain consisting of a central polar residue and multiple hydrophobic layers. This illlustration represents a typical SNARE domain. The hydrophobic residues are located in the A and D positions of the heptad repeats. The 0-layer is at position D, which is occupied by a polar or charged residue (usually glutamine or arginine). Layers are numbered according to their location relative to the 0-layer and are positive if counting towards the C-terminus and negative if counting towards the N-terminus. (B) Schematic of IncA, SNARE-like bacterial proteins found in C. trachomatis (CtrIncA) and C. caviae (CcaIncA). Residue numbers approximate the boundaries of each domain. The length of each domain is represented relative to length of the individual protein. Note that CcaIncA is actually 82 residues longer than CtrIncA. TMD=Transmembrane domain.
IncA from C. trachomatis and C. caviae encodes SNARE-like motifs
The severe loss of homeostatic regulation of vesicular trafficking in Chlamydia-infected cells and the aberrant (and Chlamydia-specific) translocation of proteins to the inclusion membrane suggest that bacterial effectors actively mediate this process. Two inclusion proteins of C. trachomatis and one protein from C. caviae show surprising structural homology to eukaryotic SNARE proteins. C. trachomatis IncA (herafter referred to as CtrIncA) is a ~34kDa protein composed of 273 amino acids. It contains the characteristic N- terminal bi-lobed transmembrane domain (TMD) altogether spanning approximately 48 amino acids (Figure 5B and Figure 2). Computational analysis determined that the TMD likely contains alpha helices [Dehoux et al., 2011]. It is unclear whether this hydrophobic stretch constitutes one long TMD or two shorter transmembrane domains separated by a cytosol-exposed linker region. In either case, the regions flanking the TMD are exposed to the cytoplasm. It is currently unknown what, if any, role the N-terminal cytosolic domain plays in pathogenesis since no conserved functional domains have been identified in this region. On the other hand, the C-terminal cytosolic region is significantly longer and contains two SNARE-like domains shown to be involved in corruption of host vesicle trafficking [Delevoye et al., 2004; Delevoye et al., 2008; Paumet et al., 2009]. It is interesting to note that the distance between the two SNARE-like domains of CtrIncA (59 amino acids) closely approximates the distance between the two SNARE domains of SNAP-25 (62 amino acids)—a SNARE protein containing two light chains and involved in neuronal regulated exocytosis. This suggests that the two SNARE-like domains may be able to physically interact with each other in a parallel coiled-coil fashion as is the case with SNAP-25 [Sutton et al., 1998]. To date, no post-translational modifications have been associated with CtrIncA.
Chlamydophila caviae IncA (hereafter referred to as CcaIncA) shares many of the same biochemical properties as CtrIncA including the presence of an N-terminal bi-lobed TMD, a cytoplasmic N-terminal domain, and two C-terminal SNARE-like domains separated by 48 amino acids [Delevoye et al., 2004] (Figure 5B). CcaIncA is slightly larger than CtrIncA at 355 amino acids and has an expected molecular weight of about 39kDa. As discussed above, CcaIncA is post-translationally modified by phosphorylation of serine 17 by an as-yet- unidentified kinase [Alzhanov et al., 2004]. There is evidence that both CtrIncA and CcaIncA are able to form homodimers and homotetramers in vivo [Delevoye et al., 2004]. It is currently hypothesized that the coiled-coil/SNARE-like domains regulate multimerization, but the nature of this interaction and its significance is poorly understood.
The hypothesis that IncA plays a role in regulating membrane fusion was first proposed when it was noticed that strains of C. trachomatis harboring mutations in their IncA gene developed inclusions that were smaller and more numerous than those of their wildtype counterparts [Suchland et al., 2000]. Whereas an infection with a wildtype strain results in an average of one large inclusion per infected cell, infection with an IncA-deficient strain results in the formation of multiple smaller inclusions that seemingly cannot fuse with one another. This homotypic fusion event seems to be important for pathogenesis because infection with strains of C. trachomatis that form non-fusogenic inclusions typically results in the manifestation of sub-clinical symptoms associated with Chlamydia [Geisler et al., 2001]. Supporting these observations, the microinjection of anti-IncA antibodies into the cytosol of infected cells replicated the non-fusogenic phenotype [Hackstadt et al., 1999] This is in agreement with a role of IncA in mediating homotypic fusion. In a yeast two-hybrid assay, CtrIncA, but not IncA from Chlamydophila psittaci, interacted with itself [Hackstadt et al., 1999]. This is important because C. psittaci infections naturally produce non-fusogenic inclusions despite having an apparently intact IncA gene. It is possible that C. psittaci IncA lacks a functional homodimerization domain.
The discovery that both CtrIncA and CcaIncA possess coiled-coil/SNARE-like domains prompted a detailed investigation of the interactions between IncA and SNARE proteins. When overexpressed in HeLa cells, CtrIncA binds to the v-SNAREs VAMP3, VAMP7, and VAMP8. This binding was confirmed in vitro for VAMP3 and VAMP8 [Delevoye et al., 2008]. The interaction between IncA and VAMP7 is dependent on the presence of the SNARE motif of VAMP7, since the deletion of this domain ablated inter-protein binding. If glutamine Q244 of CtrIncA is mutated to an arginine, the in vivo interaction with the host SNAREs is lost. VAMP3, VAMP7, and VAMP8 are v-SNAREs that contain an arginine at their zero-layer. These observations led to the speculation that the glutamine residue of IncA coordinates with the arginines of the VAMPs to facilitate binding as was observed in typical SNARE complex formation [Delevoye et al., 2008]. It remains to be seen if this property of binding to SNAREs is conserved across Chlamydiae.
Another C. trachomatis protein, CT813, was found to have at least one putative coiled-coil domain in its C-terminal cytoplasmic region [Delevoye et al., 2008]. CT813 has only been shown to bind VAMP7 and, like IncA, this interaction was dependent on the presence of VAMP7 SNARE domain. No interaction between VAMP3, VAMP4, or VAMP8 was observed. CT813 has a putative length of 264 amino acids and a molecular weight of ~29kDa. Similar to CtrIncA, it is predicted to encode a heptad repeat and contains a bi-lobed TMD. Whether CT813 has the capability to inhibit SNARE-mediated membrane fusion is not yet known.
As more laboratories unravel the topology of the inclusion membrane, we can begin to learn more about the structure of the individual proteins themselves. Of the 59 putative C. trachomatis inclusion proteins analyzed computationally by Dehoux et al., 10 (16.9%) were noted to have significant coiled-coil potential. Likewise, about 20% of the putative inclusion proteins of C. pneumoniae are predicted to form coiled-coils [Dehoux et al., 2011]. Discovering binding partners for all of these proteins would greatly enhance our knowledge of Chlamydia infections and possibly reveal novel methods used by other intracellular bacteria to corrupt host resources for their own survival.
The in vitro liposome fusion assay: A biochemical approach to a biological problem The inability to genetically manipulate Chlamydiae has proven to be a great challenge in the field. Scientists have developed novel techniques to circumvent this problem. One way to study Chlamydia proteins is by the directed mutagenesis of those proteins for use in in vitro assays. Our laboratory for example, has capitalized on a liposome fusion assay commonly used to study the biophysics of SNARE-mediated membrane fusion [Paumet et al., 2005; Varlamov et al., 2004; Weber et al., 1998].
The in vitro liposome fusion assay is a robust system for studying SNARE complex formation and functionality in a cell-free environment. The assay allows for the reduction of the membrane fusion machinery to its core components so that the contributions of mutations, effectors, or bioactive compounds can be studied. Although variations of this assay exist, they all follow the same basic workflow. Full-length SNARE proteins are first purified using standard protein expression and purification techniques. Next, each SNARE is reconstituted into either a t- or v-SNARE liposome population depending on the protein in question. Both liposome populations are of the same composition, except that the v- SNARE liposomes contain NBD and Rhodamine, a FRET (Fluorescence Resonance Energy Transfer) pair in which one fluorophore (Rhodamine) quenches the other (NBD) (Figure 6A). After purifying and recovering the liposomes on a density gradient, both populations of liposomes are mixed at 37°C. As the liposomes fuse, the distance between the FRET pair increases, and the quenching of NBD decreases. The rate of fusion is measured as an increase in NBD fluorescence (Figure 6B). The main advantage of this assay comes from the fact that one can insert virtually any membrane-bound proteins into the liposome and study their effects on SNARE-mediated membrane fusion. Due to a lack of genetic tools for manipulating the Chlamydia inclusion, this technique may prove critical for our understanding of inclusion membrane dynamics and the function of IncA and other related bacterial effectors on membrane fusion. In fact, this assay has now been successfully used to study the function of bacterial effectors on SNARE-mediated membrane fusion [Paumet et al., 2009].

Fig. 6. Results of a Typical Liposome Fusion Assay Involving IncA. (A) t-SNARE and v- SNARE liposome populations are incubated together at 37°. To test the effect of IncA on SNARE-mediated membrane fusion, we created liposomes that contained IncA in addition to the v-SNARE. As a control, v-SNARE liposomes were created without IncA. Cognate SNARE interactions result in liposome fusion and dequenching of the FRET pair. (B) Percent of maximum fluorescence is plotted as a function of time. The t-SNARE liposomes contain Syntaxins 7 and 8 and Vti1b. v-SNARE liposomes contain either VAMP8 alone (-IncA) or VAMP8 plus IncA (+IncA). The presence of IncA on the v-SNARE liposome results in an inhibition of fusion as shown by a decrease in percent maximal fluorescence.
CtrIncA and CcaIncA inhibit SNARE-mediated endocytic fusion
The late endocytic/lysosomal SNARE complex is composed of the three t-SNAREs Syntaxin 7, Syntaxin 8 and Vti1b, and the v-SNARE VAMP8 [Antonin et al., 2000; Mullock et al., 2000]. During Chlamydia infection, VAMP8 has been shown to traffic to the inclusion membrane [Delevoye et al., 2008]. That IncA interacted with VAMP8 in vitro prompted us to investigate the role IncA played in altering endocytic membrane fusion. Using the in vitro liposome fusion assay described previously, our laboratory demonstrated that both CtrIncA and CcaIncA were capable of inhibiting endocytic fusion mediated by Syn7/Syn8/Vti1b (t- SNARE) and its cognate v-SNARE, VAMP8 [Paumet et al., 2009] (Figure 6B). The location of IncA did not alter its ability to inhibit fusion since co-incubation of IncA with either the t- or v-SNARE resulted in an inhibition of fusion. This suggests that in vivo, IncA may be able to inhibit lysosomal fusion before VAMP8 is recruited to the inclusion. Using truncated mutants of IncA, our group determined that the N-terminal SNARE-like domain of either CtrIncA or CcaIncA is sufficient for inhibition. The role of the C-terminal SNARE-like domain remains to be determined.
To validate the inhibitory capacity of IncA in a cellular context, RBL-2H3 (rat basophilic leukemia) cells were transfected with myc-tagged CcaIncA containing a premature stop codon before the C-terminal SNARE-like domain [Paumet et al., 2009]. We used RBL-2H3 cells because their secretory granules are lysosome-like and contain enzymes such as b- hexosaminidase. During stimulation with PMA (phorbol-12-myristate 13- acetate)/Ionomycin, b-hexosaminidase is released into the extracellular medium in a VAMP8-dependent manner [Paumet et al., 2000; Sander et al., 2008]. The amount of b- hexosaminidase present in the supernatant after stimulation can be used as a proxy for the capacity of the lysosomal granules to fuse with the plasma membrane. Myc-tagged CcaIncA co-localized with the secretory lysosomes in RBL cells. After stimulation, we observed a significant reduction in the level of b-hexosaminidase in the supernatant. This illustrates that under physiological conditions, CcaIncA is able to inhibit the fusion of intracellular membranes. Figure 7 is a graphical representation of the proposed model for IncA-mediated membrane fusion inhibition [Wesolowski and Paumet, 2010].

Fig. 7. Proposed Model of IncA-Mediated Inhibition of Late Endocytic Fusion with the Inclusion. (Left) The fusion of endocytic compartments (EE and LE above) leads to the acidification of the phagosome and destruction of bacteria. This process is mediated by SNARE proteins, namely Syntaxin 7, Syntaxin 8, Vti1b and VAMP8. (Right) During Chlamydia infection, IncA is expressed on the surface of the inclusion where it interferes with the endocytic SNAREs and blocks membrane fusion with the lysosomes. As a result, Chlamydia inclusion avoids the degradative pathway.
8. IncA and CT813 from C. trachomatis elicit antibody responses in humans
There is currently no vaccine available for Chlamydia infections. The challenge has been to find a phylogenetically conserved Chlamydia protein capable of producing a potent and
prolonged immunological response. Finding the correct model organism in which to study this disease has also been a challenge. Mice are frequently used in immunology studies but are not the natural hosts of C. trachomatis, the causative agent of many human illnesses. Instead, mice are host to the related C. muridarum (also called Mouse Pneumonitis strain, or MoPn). While the use of an animal model has greatly facilitated the study of the pathogenesis of Chlamydiae infections [Brunham and Rey-Ladino, 2005], differences between the mouse and human etiologic agents of disease makes translation of results between both organisms inherently difficult to interpret. Nevertheless, progress is being made in the identification of immunogenic Chlamydia proteins in humans.
Array-based technologies have been utilized to scan immunized human sera for reactivity with C. trachomatis proteins. For example, GST-tagged Chlamydia proteins have been coated on a microtiter plate to scan the sera of infected women [Wang et al., 2010]. Antibodies in the sera bound to their respective antigens were visualized by horseradish peroxidase-based ELISA. Of the 933 ORFs tested, 719 were antigenic (bound to human anti-Chlamydia antibodies). The antigens that were recognized by 50% or greater of the patients from whom the sera were taken were designated as “immunodominant antigens” [Wang et al., 2010]. The number of immunodominant antigens totaled 26. Included in those 26 antigens were IncA (also known as CT119 for C. trachomatis IncA) and CT813—two proteins with known SNARE protein-binding capacity. These proteins were recognized by 65% and 79% of the sera tested, respectively. Notably, 6 of the 26 identified antigens were previously shown by the same group to localize to the inclusion membrane [Li et al., 2008]. CT089, CT119 (IncA), CT147, CT442, CT529, and CT813 all elicited antibody responses from more than 65% of infected women and localize to the inclusion membrane.
Antigens were further classified as being either infection-dependent or infection- independent based on their reactivity with serum from mice and rabbits. Briefly, animals were either infected with live C. trachomatis serovar D or immunized with UV-killed bacteria and their sera collected after booster infections or immunizations [Wang et al., 2010]. The antigens recognized by immunized sera were designated “infection-independent” while the rest were “infection-dependent.”5 CT089, CT442, CT529, and CT813 were determined to be infection-dependent while immunized sera produced from inoculation with dead bacteria recognized IncA and CT147 [Wang et al., 2010].
Interestingly, using a similar approach, Cruz-Fisher et al. found that antibodies against C. muridarum IncA (also known as TC0396) were only produced in mice infected with live elementary bodies not UV-killed organisms [Cruz-Fisher et al., 2011]. The antigenicity was not dependent on the route of infection as either intranasal or intravaginal inoculations resulted in the induction of a robust humoral response evidenced by the production of anti- IncA (anti-TC0396) antibodies. The different requirements for production of antibody by live versus dead inoculations between the two Chlamydia species may reflect a difference in antigen processing and presentation between mice and humans. It could also be the result of differences between the pathology of C. muridarum and that of C. trachomatis.
It is intriguing that some inclusion proteins appear to be highly immunogenic in humans, hinting at possible targets for future vaccine development6 [Li et al., 2007]. This observation makes studying the biochemical properties of inclusion proteins all the more important. Uncovering the physiologic role(s) of these bacterial effectors could greatly advance our ability to engineer immunogenic epitopes with minimal side effects for use in vaccine studies.
Understanding more about the immunology of Chlamydia infections will lead to the design of novel vaccine strategies to combat the threat of sexually transmitted infections and the resulting medical complications. The discovery of highly antigenic bacterial effectors is a promising start. One challenge that remains is defining the immunogenic epitopes on each of these so-called “immunodominant antigens.” By narrowing down the sites of antibody binding, it may be possible to design synthetic peptides that protect against a broad range of C. trachomatis serovars and elicit long-lasting cellular immunity.
9. Future research
We are still far from providing a comprehensive picture of the molecular mechanisms of Chlamydia infection. High-throughput functional assays could reveal a wealth of information about Chlamydia’s ability to corrupt host signaling and trafficking pathways. However, the lack of a genetic system has been a barrier to the development of those assays that could potentially identify novel Chlamydia-specific antibiotic targets. One laboratory recently reported the generation of point mutations in C. trachomatis after treatment with ethyl methanesulfonate [Kari et al., 2011]. This group successfully isolated a clone mutated for the trpB tryptophan synthase gene, but otherwise isogenic to the wildtype parent strain. The isolation and identification of mutants, however, is labor-intensive and may not be amenable to large-scale genetic screens in most laboratories. However, this represents an important step in beginning to manipulate the Chlamydia genome.
The function of inclusion membrane proteins needs to be explored more in depth. Their location at the host-pathogen interface makes them ideal candidates for virulence factors and also as druggable targets for novel therapeutics. Despite years of research on Chlamydia, little is known about the role of inclusion proteins in the etiology of infection. The best- characterized inclusion protein is IncA. Although a role for IncA in promoting homotypic inclusion fusion has been known for over ten years [Hackstadt et al., 1999], only recently has the interaction between the host endocytic SNARE complex and IncA been explored in detail. That other Inc proteins also contain putative coiled-coil domains strongly suggests that they may interact with cytosol-exposed host proteins including SNAREs. These interactions between Chlamydia and host proteins could potentially explain many of the observations noted by others throughout the years such as the homotypic fusion of inclusions, inhibition of fusion with late endosomes/lysosomes, or the acquisition of Golgi- associated membrane markers.
Despite the lack of a tractable genetic system for studying Chlamydia, new and important discoveries are still being made. In particular, the cell biology of Chlamydia infection has revealed how intracellular infection can alter host vesicle trafficking. The availability of compartment-specific markers and fluorescent probes has greatly accelerated our understanding of the re-localization of host proteins and lipids during infection. The study regarding sphingomyelin is one example of how the use of cell biology techniques has overcome the inherent problems that come with working with a genetically recalcitrant bacterium. In order to continue to study the molecular mechanisms of Chlamydia infection, new techniques will have to be developed.
10. Conclusion
Despite strong efforts to combat Chlamydia, it continues to be the most frequently reported bacterial STD in the United States. Although Chlamydia is commonly associated with venereal diseases, infection also occurs in the eyes or lungs. Infection of tissues surrounding the eye can result in a form of infectious blindness known as trachoma—a widespread problem in regions of Africa, South America, and Asia (data from World Health Organization, Global Health Observatory). Chlamydia pneumoniae can colonize lung tissue and is the causative agent of a form of bacteria-induced pneumonia. Recent studies have also attributed increased risk of heart disease—including atherosclerosis—to Chlamydia infection [Beagley et al., 2009].
The normal course of treatment for Chlamydia infections involves the use of antibiotics. However, non-specific antibiotics like the ones used to treat Chlamydia may negatively affect the mucosal microbiome that helps to regulate essential processes like digestion, inflammation and immune responses. Additionally, some antibiotics have detrimental side effects. Therefore, finding novel and specific drug targets that protect against a wide range of Chlamydia serovars is of extreme importance.
As an obligate intracellular pathogen, Chlamydiae must adapt to acquire host nutrients while avoiding destruction by innate immune cells. Through the selective interactions with Golgi- derived vesicles, lipid droplets, and multivesicular bodies, Chlamydiae obtain host nutrients such as sphingomyelin that support growth and accelerate infection. While they promote fusion with these organelles, Chlamydiae simultaneously inhibit fusion with lysosomes and avoid compartment acidification and degradation by processes that are still unclear.
Evidence suggests that Chlamydiae employ a Type 3 Secretion System to translocate a special subset of effectors known as Inc proteins to the inclusion membrane surface, and that these proteins are involved in subversion of host vesicle trafficking. The identity of these Inc proteins is still unclear. This is hampered by the genetic diversity of the bacteria themselves and a lack of genetic tools for studying Chlamydiae. Incs known to localize to the inclusion membrane share little amino acid sequence homology, but may share structural or functional similarities including the presence of a bi-lobed hydrophobic transmembrane domain and, for a number of putative Incs, coiled-coil domain(s). IncA is one such inclusion protein that is transported by a T3SS and resides on the inclusion membrane. CtrIncA has been shown to interact with host SNARE proteins VAMP3, VAMP7, and VAMP8 possibly via its coiled-coils. It also inhibits endocytic SNARE-mediated fusion in a specific manner. Another C. trachomatis protein, CT813, localizes to the inclusion membrane [Chen et al., 2006] and may also interfere with SNARE-mediated membrane fusion [Delevoye et al.,
2008]. The biophysical studies of IncA presented in this chapter begin to reconcile our understanding of host vesicle subversion with what we know about inclusion membrane composition and could lead to the future design of novel anti-Chlamydia therapeutics.
11. Acknowledgement
We would like to thank the entire Chlamydia pathogenesis community for its work that made the review presented in this chapter possible. In particular, we are grateful to the members of the Paumet laboratory for their contributions to the Chlamydia SNARE-like proteins project. We would like to apologize to those authors whose work was not cited owing to space limitations. This research is supported by the National Institutes of Health grant #RO1 AI073486 (to F.P.).
12. References
Alzhanov, D., Barnes, J., Hruby, D.E., and Rockey, D.D. (2004). Chlamydial development is blocked in host cells transfected with Chlamydophila caviae incA. BMC Microbiology 4, 24 %U http://www.ncbi.nlm.nih.gov/pubmed/15230981.
Antonin, W., Fasshauer, D., Becker, S., Jahn, R., and Schneider, T.R. (2002). Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat Struct Mol Biol 9, 107-111 %U http://dx.doi.org/110.1038/nsb1746.
Antonin, W., Holroyd, C., Tikkanen, R., Honing, S., and Jahn, R. (2000). The R-SNARE endobrevin/VAMP-8 mediates homotypic fusion of early endosomes and late endosomes. Mol Biol Cell 11, 3289-3298.
Bannantine, J.P., Griffiths, R.S., Viratyosin, W., Brown, W.J., and Rockey, D.D. (2000). A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cellular Microbiology 2, 35-47 %U http://www.ncbi.nlm.nih.gov/pubmed/11207561.
Beagley, K.W., Huston, W.M., Hansbro, P.M., and Timms, P. (2009). Chlamydial infection of immune cells: altered function and implications for disease. Crit Rev Immunol 29, 275-305.
Beatty, W.L. (2006). Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. Journal of Cell Science 119, 350-359 %U http://www.ncbi.nlm.nih.gov/pubmed/16410552.
Beatty, W.L. (2008). Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infection and Immunity 76, 2872-2881 %U http://www.ncbi.nlm.nih.gov/pubmed/18426873.
Bock, J.B., Klumperman, J., Davanger, S., and Scheller, R.H. (1997). Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Molecular biology of the cell 8, 1261-1271.
Bock, J.B., Matern, H.T., Peden, A.A., and Scheller, R.H. (2001). A genomic perspective on membrane compartment organization. Nature 409, 839-841.
Bostrom, P., Andersson, L., Rutberg, M., Perman, J., Lidberg, U., Johansson, B.R., Fernandez-Rodriguez, J., Ericson, J., Nilsson, T., Boren, J., et al. (2007). SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nature Cell Biology 9, 1286-1293 %U http://www.ncbi.nlm.nih.gov/pubmed/17922004.
Botelho, R.J., and Grinstein, S. (2011). Phagocytosis. Curr Biol 21, R533-538.
Brunham, R.C., and Rey-Ladino, J. (2005). Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol 5, 149-161.
Capmany, A., and Damiani, M.a.T. (2010). Chlamydia trachomatis intercepts Golgi-derived sphingolipids through a Rab14-mediated transport required for bacterial development and replication. PLoS One 5, e14084 %U http://www.ncbi.nlm.nih.gov/pubmed/21124879.
Chen, C., Chen, D., Sharma, J., Cheng, W., Zhong, Y., Liu, K., Jensen, J., Shain, R., Arulanandam, B., and Zhong, G. (2006). The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infection and Immunity 74, 4826-4840.
Chen, Y.A., and Scheller, R.H. (2001). SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2, 98-106.
Cocchiaro, J.L., Kumar, Y., Fischer, E.R., Hackstadt, T., and Valdivia, R.H. (2008).
Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proceedings of the National Academy of Sciences of the United States of America 105, 9379-9384.
Conibear, E., and Stevens, T.H. (1998). Multiple sorting pathways between the late Golgi and the vacuole in yeast. Biochim Biophys Acta 1404, 211-230.
Cruz-Fisher, M.I., Cheng, C., Sun, G., Pal, S., Teng, A., Molina, D.M., Kayala, M.A., Vigil, A., Baldi, P., Felgner, P.L., et al. (2011). Identification of immunodominant antigens by probing a whole Chlamydia trachomatis open reading frame proteome microarray
using sera from immunized mice. Infection and Immunity 79, 246-257.
Dehoux, P., Flores, R., Dauga, C., Zhong, G., and Subtil, A. (2011). Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: the Inc proteins. BMC Genomics 12, 109 %U
http://www.ncbi.nlm.nih.gov/pubmed/21324157.
Delevoye, C., Nilges, M., Dautry-Varsat, A., and Subtil, A. (2004). Conservation of the
biochemical properties of IncA from Chlamydia trachomatis and Chlamydia
caviae: oligomerization of IncA mediates interaction between facing membranes. J
Biol Chem 279, 46896-46906.
Delevoye, C.d., Nilges, M., Dehoux, P., Paumet, F., Perrinet, S.p., Dautry-Varsat, A., and
Subtil, A. (2008). SNARE protein mimicry by an intracellular bacterium. PLoS
Pathogens 4, e1000022 %U http://www.ncbi.nlm.nih.gov/pubmed/18369472.
Farese, R.V., Jr., and Walther, T.C. (2009). Lipid droplets finally get a little R-E-S-P-E-C-T.
Cell 139, 855-860 %U http://www.ncbi.nlm.nih.gov/pubmed/19945371.
Fasshauer, D. (2003). Structural insights into the SNARE mechanism. Biochim Biophys Acta
1641, 87-97.
Fasshauer, D., Sutton, R.B., Brunger, A.T., and Jahn, R. (1998). Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs.
Proc Natl Acad Sci U S A 95, 15781-15786.
Fields, K.A., Fischer, E., and Hackstadt, T. (2002). Inhibition of fusion of Chlamydia
trachomatis inclusions at 32 degrees C correlates with restricted export of IncA.
Infection and Immunity 70, 3816-3823 %U
http://www.ncbi.nlm.nih.gov/pubmed/12065525.
Fields, K.A., and Hackstadt, T. (2002). The chlamydial inclusion: escape from the endocytic
pathway. Annual Review of Cell and Developmental Biology 18, 221-245 %U
http://www.ncbi.nlm.nih.gov/pubmed/12142274.
Fukuda, R., McNew, J.A., Weber, T., Parlati, F., Engel, T., Nickel, W., Rothman, J.E., and Sollner, T.H. (2000). Functional architecture of an intracellular membrane t-SNARE. Nature 407, 198-202.
Geisler, W.M., Suchland, R.J., Rockey, D.D., and Stamm, W.E. (2001). Epidemiology and clinical manifestations of unique Chlamydia trachomatis isolates that occupy nonfusogenic inclusions. The Journal of Infectious Diseases 184, 879-884 %U http://www.ncbi.nlm.nih.gov/pubmed/11528595.
Hackstadt, T., Rockey, D.D., Heinzen, R.A., and Scidmore, M.A. (1996). Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. The EMBO Journal 15, 964-977 %U http://www.ncbi.nlm.nih.gov/pubmed/8605892.
Hackstadt, T., Scidmore, M.A., and Rockey, D.D. (1995). Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proceedings of the National Academy of Sciences of the United States of America 92, 4877-4881 %U http://www.ncbi.nlm.nih.gov/pubmed/7761416.
Hackstadt, T., Scidmore-Carlson, M.A., Shaw, E.I., and Fischer, E.R. (1999). The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cellular Microbiology 1, 119-130 %U http://www.ncbi.nlm.nih.gov/pubmed/11207546.
Hackstadt, T., Todd, W.J., and Caldwell, H.D. (1985). Disulfide-mediated interactions of the chlamydial major outer membrane protein: role in the differentiation of chlamydiae? Journal of Bacteriology 161, 25-31 %U http://www.ncbi.nlm.nih.gov/pubmed/2857160.
Heijnen, H.F., Schiel, A.E., Fijnheer, R., Geuze, H.J., and Sixma, J.J. (1999). Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules.
Blood 94, 3791-3799.
Heinzen, R.A., Scidmore, M.A., Rockey, D.D., and Hackstadt, T. (1996). Differential interaction
with endocytic and exocytic pathways distinguish parasitophorous vacuoles of
Coxiella burnetii and Chlamydia trachomatis. Infect Immun 64, 796-809.
Hueck, C.J. (1998). Type III protein secretion systems in bacterial pathogens of animals and
plants. Microbiol Mol Biol Rev 62, 379-433.
Hutagalung, A.H., and Novick, P.J. (2011). Role of Rab GTPases in membrane traffic and cell
physiology. Physiological Reviews 91, 119-149 %U
http://www.ncbi.nlm.nih.gov/pubmed/21248164.
Johnson, V.J., He, Q., Osuchowski, M.F., and Sharma, R.P. (2004). Disruption of sphingolipid
homeostasis by myriocin, a mycotoxin, reduces thymic and splenic T-lymphocyte
populations. Toxicology 201, 67-75 %U
http://www.ncbi.nlm.nih.gov/pubmed/15297021.
Kari, L., Goheen, M.M., Randall, L.B., Taylor, L.D., Carlson, J.H., Whitmire, W.M., Virok, D.,
Rajaram, K., Endresz, V., McClarty, G., et al. (2011). Generation of targeted
Chlamydia trachomatis null mutants. Proceedings of the National Academy of
Sciences of the United States of America 108, 7189-7193.
Kumar, Y., Cocchiaro, J., and Valdivia, R.H. (2006). The obligate intracellular pathogen
Chlamydia trachomatis targets host lipid droplets. Current Biology: CB 16, 1646-
1651 %U http://www.ncbi.nlm.nih.gov/pubmed/16920627.
Li, W., Guentzel, M.N., Seshu, J., Zhong, G., Murthy, A.K., and Arulanandam, B.P. (2007).
Induction of cross-serovar protection against genital chlamydial infection by a
targeted multisubunit vaccination approach. Clin Vaccine Immunol 14, 1537-1544.
Li, Z., Chen, C., Chen, D., Wu, Y., Zhong, Y., and Zhong, G. (2008). Characterization of fifty
putative inclusion membrane proteins encoded in the Chlamydia trachomatis
genome. Infection and Immunity 76, 2746-2757 %U
http://www.ncbi.nlm.nih.gov/pubmed/18391011.
McNew, J.A., Parlati, F., Fukuda, R., Johnston, R.J., Paz, K., Paumet, F., Sollner, T.H., and
Rothman, J.E. (2000). Compartmental specificity of cellular membrane fusion
encoded in SNARE proteins. Nature 407, 153-159.
Melia, T.J., Weber, T., McNew, J.A., Fisher, L.E., Johnston, R.J., Parlati, F., Mahal, L.K.,
Sollner, T.H., and Rothman, J.E. (2002). Regulation of membrane fusion by the
membrane-proximal coil of the t-SNARE during zippering of SNAREpins. The
Journal of Cell Biology 158, 929-940.
Metchnikoff, E. (1891). Lecture on Phagocytosis and Immunity. Br Med J 1, 213-217.
Moore, E.R., Mead, D.J., Dooley, C.A., Sager, J., and Hackstadt, T. (2011). The trans-Golgi
SNARE syntaxin 6 is recruited to the chlamydial inclusion membrane.
Microbiology 157, 830-838.
Morrison, D.K. (2009). The 14-3-3 proteins: integrators of diverse signaling cues that impact
cell fate and cancer development. Trends Cell Biol 19, 16-23.
Mullock, B.M., Smith, C.W., Ihrke, G., Bright, N.A., Lindsay, M., Parkinson, E.J., Brooks,
D.A., Parton, R.G., James, D.E., Luzio, J.P., et al. (2000). Syntaxin 7 is localized to late endosome compartments, associates with Vamp 8, and Is required for late endosome-lysosome fusion. Mol Biol Cell 11, 3137-3153.
Muschiol, S., Bailey, L., Gylfe, A., Sundin, C., Hultenby, K., Bergström, S., Elofsson, M., Wolf-Watz, H., Normark, S., and Henriques-Normark, B. (2006). A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proceedings of the National Academy of Sciences of the United States of America 103, 14566-14571 %U
http://www.ncbi.nlm.nih.gov/pubmed/16973741.
Paavonen, J., and Eggert-Kruse, W. (1999). Chlamydia trachomatis: impact on human
reproduction. Hum Reprod Update 5, 433-447.
Parlati, F., Varlamov, O., Paz, K., McNew, J.A., Hurtado, D., Sollner, T.H., and Rothman, J.E.
(2002). Distinct SNARE complexes mediating membrane fusion in Golgi transport
based on combinatorial specificity. Proceedings of the National Academy of
Sciences of the United States of America 99, 5424-5429.
Paumet, F., Brugger, B., Parlati, F., McNew, J.A., Sollner, T.H., and Rothman, J.E. (2001). A t-SNARE of the endocytic pathway must be activated for fusion. The Journal of Cell Biology 155, 961-968.
Paumet, F., Le Mao, J., Martin, S., Galli, T., David, B., Blank, U., and Roa, M. (2000). Soluble NSF attachment protein receptors (SNAREs) in RBL-2H3 mast cells: functional role
of syntaxin 4 in exocytosis and identification of a vesicle-associated membrane
protein 8-containing secretory compartment. J Immunol 164, 5850-5857.
Paumet, F., Rahimian, V., Di Liberto, M., and Rothman, J.E. (2005). Concerted auto- regulation in yeast endosomal t-SNAREs. The Journal of Biological Chemistry 280, 21137-21143.
Paumet, F., Rahimian, V., and Rothman, J.E. (2004). The specificity of SNARE-dependent fusion is encoded in the SNARE motif. Proceedings of the National Academy of Sciences of the United States of America 101, 3376-3380.
Paumet, F., Wesolowski, J., Garcia-Diaz, A., Delevoye, C., Aulner, N., Shuman, H.A., Subtil, A., and Rothman, J.E. (2009). Intracellular bacteria encode inhibitory SNARE-like proteins. PLoS One 4, e7375.
Pelham, H.R. (1999). SNAREs and the secretory pathway-lessons from yeast. Exp Cell Res 247, 1-8.
Peters, J., Wilson, D., Myers, G., Timms, P., and Bavoil, P. (2007). Type III secretion a la Chlamydia. Trends in Microbiology 15, 241-251.
Pfeffer, S.R. (2001). Rab GTPases: specifying and deciphering organelle identity and function. Trends Cell Biol 11, 487-491.
Pobbati, A.V., Stein, A., and Fasshauer, D. (2006). N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science 313, 673-676.
Robertson, D.K., Gu, L., Rowe, R.K., and Beatty, W.L. (2009). Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathogens 5, e1000664.
Rockey, D.D., Grosenbach, D., Hruby, D.E., Peacock, M.G., Heinzen, R.A., and Hackstadt, T.
(1997). Chlamydia psittaci IncA is phosphorylated by the host cell and is exposed on the cytoplasmic face of the developing inclusion. Molecular Microbiology 24, 217-228 %U http://www.ncbi.nlm.nih.gov/pubmed/9140978.
Rockey, D.D., Heinzen, R.A., and Hackstadt, T. (1995). Cloning and characterization of a
Chlamydia psittaci gene coding for a protein localized in the inclusion membrane
of infected cells. Molecular Microbiology 15, 617-626 %U http://www.ncbi.nlm.nih.gov/pubmed/7783634.
Rzomp, K.A., Scholtes, L.D., Briggs, B.J., Whittaker, G.R., and Scidmore, M.A. (2003). Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infection and Immunity 71, 5855-5870 %U http://www.ncbi.nlm.nih.gov/pubmed/14500507.
Sander, L.E., Frank, S.P., Bolat, S., Blank, U., Galli, T., Bigalke, H., Bischoff, S.C., and Lorentz, A. (2008). Vesicle associated membrane protein (VAMP)-7 and VAMP-8, but not VAMP-2 or VAMP-3, are required for activation-induced degranulation of mature
human mast cells. Eur J Immunol 38, 855-863.
Schramm, N., Bagnell, C.R., and Wyrick, P.B. (1996). Vesicles containing Chlamydia trachomatis serovar L2 remain above pH 6 within HEC-1B cells. Infection and Immunity 64, 1208-1214.
Schroeder, J.J., Crane, H.M., Xia, J., Liotta, D.C., and Merrill, A.H. (1994). Disruption of sphingolipid metabolism and stimulation of DNA synthesis by fumonisin B1. A
molecular mechanism for carcinogenesis associated with Fusarium moniliforme.
Journal of Biological Chemistry 269, 3475 -3481 %U http://www.jbc.org/content/3269/3475/3475.abstract.
Scidmore, M.A., and Hackstadt, T. (2001). Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG.
Molecular Microbiology 39, 1638-1650 %U
http://www.ncbi.nlm.nih.gov/pubmed/11260479.
Segev, N. (2001). Ypt/rab gtpases: regulators of protein trafficking. Sci STKE 2001, re11.
Stein, A., Weber, G., Wahl, M.C., and Jahn, R. (2009). Helical extension of the neuronal SNARE complex into the membrane. Nature 460, 525-528 %U http://www.ncbi.nlm.nih.gov/pubmed/19571812.
Stephens, R.S., Kalman, S., Lammel, C., Fan, J., Marathe, R., Aravind, L., Mitchell, W., Olinger, L., Tatusov, R.L., Zhao, Q., et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science (New York, NY) 282, 754-759 %U http://www.ncbi.nlm.nih.gov/pubmed/9784136.
Stewardson, A.J., Huttner, B., and Harbarth, S. (2011). At least it won't hurt: the personal risks of antibiotic exposure. Curr Opin Pharmacol.
Subtil, A., Delevoye, C., Balana, M.E., Tastevin, L., Perrinet, S., and Dautry-Varsat, A. (2005).
A directed screen for chlamydial proteins secreted by a type III mechanism identifies a translocated protein and numerous other new candidates. Molecular Microbiology 56, 1636-1647.
Subtil, A., Parsot, C., and Dautry-Varsat, A. (2001). Secretion of predicted Inc proteins of Chlamydia pneumoniae by a heterologous type III machinery. Molecular Microbiology 39, 792-800 %U http://www.ncbi.nlm.nih.gov/pubmed/11169118.
Suchland, R.J., Rockey, D.D., Bannantine, J.P., and Stamm, W.E. (2000). Isolates of Chlamydia trachomatis that occupy nonfusogenic inclusions lack IncA, a protein localized to the inclusion membrane. Infection and Immunity 68, 360-367 %U http://www.ncbi.nlm.nih.gov/pubmed/10603409.
Sutton, R.B., Fasshauer, D., Jahn, R., and Brunger, A.T. (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347-353 %U http://www.ncbi.nlm.nih.gov/pubmed/9759724.
Tamura, A., Matsumoto, A., Manire, G.P., and Higashi, N. (1971). Electron microscopic observations on the structure of the envelopes of mature elementary bodies and developmental reticulate forms of Chlamydia psittaci. Journal of Bacteriology 105,
355-360.
Todd, W.J., and Storz, J. (1975). Ultrastructural cytochemical evidence for the activation of lysosomes in the cytocidal effect of Chlamydia psittaci. Infect Immun 12, 638-646.
van Ooij, C., Apodaca, G., and Engel, J. (1997). Characterization of the Chlamydia trachomatis vacuole and its interaction with the host endocytic pathway in HeLa cells. Infect Immun 65, 758-766.
Varlamov, O., Volchuk, A., Rahimian, V., Doege, C.A., Paumet, F., Eng, W.S., Arango, N., Parlati, F., Ravazzola, M., Orci, L., et al. (2004). i-SNAREs: inhibitory SNAREs that fine-tune the specificity of membrane fusion. The Journal of Cell Biology 164, 79-88 %U http://www.ncbi.nlm.nih.gov/pubmed/14699088.
Wang, J., Zhang, Y., Lu, C., Lei, L., Yu, P., and Zhong, G. (2010). A genome-wide profiling of the humoral immune response to Chlamydia trachomatis infection reveals vaccine candidate antigens expressed in humans. Journal of immunology 185, 1670-1680.
Watson, R.T., and Pessin, J.E. (2000). Functional cooperation of two independent targeting domains in syntaxin 6 is required for its efficient localization in the trans-golgi network of 3T3L1 adipocytes. The Journal of Biological Chemistry 275, 1261-1268.
Weber, T., Zemelman, B.V., McNew, J.A., Westermann, B., Gmachl, M., Parlati, F., Sollner, T.H., and Rothman, J.E. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759-772.
Wesolowski, J., and Paumet, F. (2010). SNARE motif: a common motif used by pathogens to manipulate membrane fusion. Virulence 1, 319-324.
Worrall, L.J., Lameignere, E., and Strynadka, N.C. (2011). Structural overview of the
bacterial injectisome. Curr Opin Microbiol 14, 3-8.
Yeung, T., Heit, B., Dubuisson, J.F., Fairn, G.D., Chiu, B., Inman, R., Kapus, A., Swanson, M., and Grinstein, S. (2009). Contribution of phosphatidylserine to membrane surface charge and protein targeting during phagosome maturation. The Journal of Cell Biology 185, 917-928.

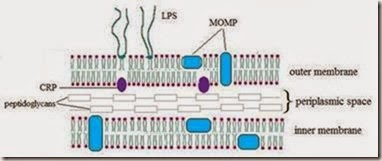
Comments
Post a Comment